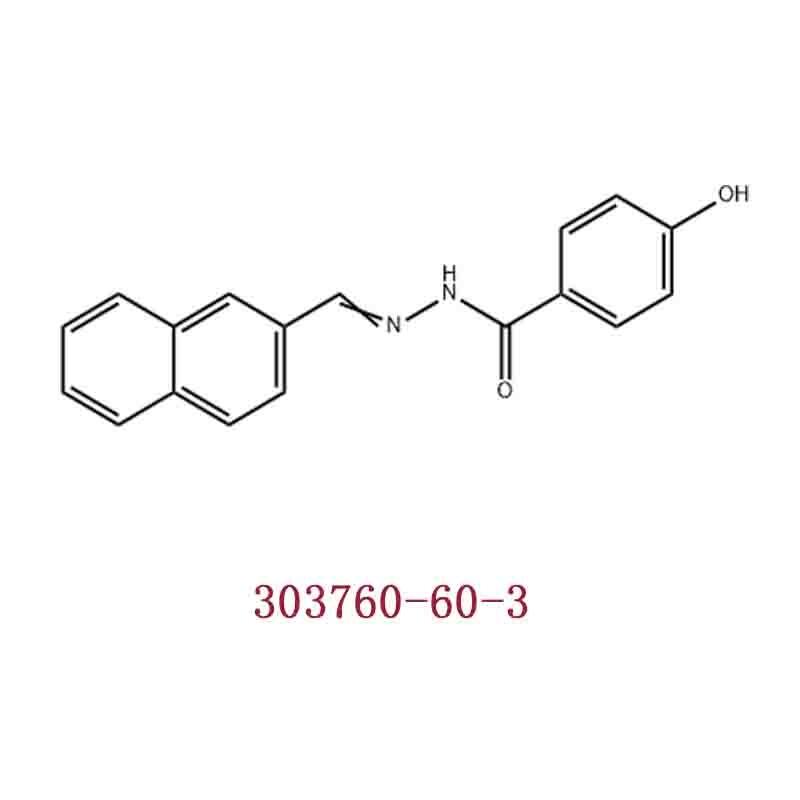
-
Categories
-
Pharmaceutical Intermediates
-
Active Pharmaceutical Ingredients
-
Food Additives
- Industrial Coatings
- Agrochemicals
- Dyes and Pigments
- Surfactant
- Flavors and Fragrances
- Chemical Reagents
- Catalyst and Auxiliary
- Natural Products
- Inorganic Chemistry
-
Organic Chemistry
-
Biochemical Engineering
- Analytical Chemistry
-
Cosmetic Ingredient
- Water Treatment Chemical
-
Pharmaceutical Intermediates
Promotion
ECHEMI Mall
Wholesale
Weekly Price
Exhibition
News
-
Trade Service
iNature Wilson's disease (WD, also known as hepatolenticular degeneration) is a rare genetic disorder that results in pathological copper storage primarily in the liver and nervous system due to mutations in the ATP7B gene
.
Hepatocyte transplantation shows therapeutic potential, however, this strategy is often hampered by shortages of high-quality donor cells and alloimmune rejection
.
On March 27, 2022, Wang Xiaoping's team from Shanghai Jiao Tong University published a research paper online in Hepatology (IF=17) entitled "ATP7B gene therapy of autologous reprogrammed hepatocytes alleviates copper accumulation in a murine model of Wilson's disease".
Small molecules reprogrammed hepatocytes from ATP7B −/− mice to harvest sufficient hepatic progenitor cells (LPCs), and these small molecules exhibited strong proliferative and hepatic differentiation abilities in vitro
.
Functional LPC-ATP7B-Heps were developed following lentivirus-mediated transfection and redifferentiation of the miniATP7B gene
.
RNA-seq data showed that compared with LPC-GFP-Heps, LPC-ATP7B-Heps gene enrichment was mainly concentrated in oxidative stress and apoptosis pathways, and copper ions in LPC-ATP7B-Heps under high copper stress Binding and cell proliferation pathway enrichment
.
Transplantation of LPC-ATP7B-Heps into ATP7B−/− mice attenuated excess hepatic copper deposition and its associated inflammation and fibrosis, which was comparable to that observed using normal primary hepatocytes four months after transplantation
.
In conclusion, this study established a new autologous reprogrammed WD hepatocyte system and achieved ATP7B gene therapy in vitro
.
LPC-ATP7B-Heps transplantation demonstrated therapeutic effects on copper homeostasis in a WD mouse model
.
Wilson disease (WD), also known as hepatolenticular degeneration, is a congenital disorder associated with a recessive pathogenic variant in the ATP7B gene, a copper-transporting ATPase expressed primarily in hepatocytes
.
Impaired copper transport to bile in WD results in a pathological accumulation of copper in the liver and then in the brain
.
Early disease detection, identification, and treatment can prevent the progression of WD
.
Pharmacological interventions (eg, zinc salts and copper chelators) reduce pathological copper deposition but do not restore normal copper metabolism
.
Orthotopic liver transplantation is an effective treatment for WD, but the shortage of donor organs and the need for lifelong immunosuppression limit its application
.
Therefore, there is an urgent need for alternative therapies focused on restoring copper homeostasis in affected WD patients
.
Cell-based therapy has emerged as an alternative approach to rescue metabolic liver disease
.
When endogenous cells are destroyed, transplanted hepatocytes survive and proliferate vigorously, repopulating the liver and performing normal liver function
.
Primary hepatocytes (PHs) from unaffected individuals are considered the optimal cell type for transplantation, as demonstrated by the effects of delivering normal rat PHs into LEC rats, a rodent model of WD eliminates liver copper and reversed the disease
.
Despite its therapeutic potential, PH cannot proliferate in vitro, and freshly isolated PH rapidly loses its hepatic gene expression profile
.
In view of this, several studies were performed focusing on in vitro hepatocyte expansion
.
Previous reports have also shown that, upon induction by small molecules, mouse and human PH can be transformed into tubular hepatic progenitor cells (LPCs) that exhibit the potential for cell proliferation and differentiation into functional hepatocytes
.
However, some disadvantages remain, including immune responses between allografts and ethical concerns
.
Schematic diagram of the article (figure from Hepatology) Therefore, gene therapy of autologous reprogrammed hepatocytes from WD animals or patients becomes a potential option
.
Previous data demonstrated the feasibility of lentivirus-mediated ATP7B overexpression in iPSC-differentiated HLC from WD patients or ATP7B gene transfer into bone marrow mesenchymal stem cells (BM-MSCs) from LEC rats
.
Both have been shown to reduce copper load in cells or in rat livers
.
To achieve this, PH from ATP7B −/− mice was reprogrammed to generate LPC by modifying an earlier established reprogramming protocol
.
This study found that hepatocyte reprogramming and expansion medium (HREM) significantly promoted LPC expansion to more than 40 passages
.
Following ATP7B overexpression, redifferentiated LPC-ATP7B-Heps were able to repopulate the livers of ATP7B −/− mice and reduce hepatic copper accumulation
.
Thus, LPC-ATP7B-Heps show therapeutic potential, and the results could pave the way for the development of new treatments for WD and similar genetic diseases of the liver
.
Reference message: https://aasldpubs.
onlinelibrary.
wiley.
com/doi/10.
1002/hep.
32484
.
Hepatocyte transplantation shows therapeutic potential, however, this strategy is often hampered by shortages of high-quality donor cells and alloimmune rejection
.
On March 27, 2022, Wang Xiaoping's team from Shanghai Jiao Tong University published a research paper online in Hepatology (IF=17) entitled "ATP7B gene therapy of autologous reprogrammed hepatocytes alleviates copper accumulation in a murine model of Wilson's disease".
Small molecules reprogrammed hepatocytes from ATP7B −/− mice to harvest sufficient hepatic progenitor cells (LPCs), and these small molecules exhibited strong proliferative and hepatic differentiation abilities in vitro
.
Functional LPC-ATP7B-Heps were developed following lentivirus-mediated transfection and redifferentiation of the miniATP7B gene
.
RNA-seq data showed that compared with LPC-GFP-Heps, LPC-ATP7B-Heps gene enrichment was mainly concentrated in oxidative stress and apoptosis pathways, and copper ions in LPC-ATP7B-Heps under high copper stress Binding and cell proliferation pathway enrichment
.
Transplantation of LPC-ATP7B-Heps into ATP7B−/− mice attenuated excess hepatic copper deposition and its associated inflammation and fibrosis, which was comparable to that observed using normal primary hepatocytes four months after transplantation
.
In conclusion, this study established a new autologous reprogrammed WD hepatocyte system and achieved ATP7B gene therapy in vitro
.
LPC-ATP7B-Heps transplantation demonstrated therapeutic effects on copper homeostasis in a WD mouse model
.
Wilson disease (WD), also known as hepatolenticular degeneration, is a congenital disorder associated with a recessive pathogenic variant in the ATP7B gene, a copper-transporting ATPase expressed primarily in hepatocytes
.
Impaired copper transport to bile in WD results in a pathological accumulation of copper in the liver and then in the brain
.
Early disease detection, identification, and treatment can prevent the progression of WD
.
Pharmacological interventions (eg, zinc salts and copper chelators) reduce pathological copper deposition but do not restore normal copper metabolism
.
Orthotopic liver transplantation is an effective treatment for WD, but the shortage of donor organs and the need for lifelong immunosuppression limit its application
.
Therefore, there is an urgent need for alternative therapies focused on restoring copper homeostasis in affected WD patients
.
Cell-based therapy has emerged as an alternative approach to rescue metabolic liver disease
.
When endogenous cells are destroyed, transplanted hepatocytes survive and proliferate vigorously, repopulating the liver and performing normal liver function
.
Primary hepatocytes (PHs) from unaffected individuals are considered the optimal cell type for transplantation, as demonstrated by the effects of delivering normal rat PHs into LEC rats, a rodent model of WD eliminates liver copper and reversed the disease
.
Despite its therapeutic potential, PH cannot proliferate in vitro, and freshly isolated PH rapidly loses its hepatic gene expression profile
.
In view of this, several studies were performed focusing on in vitro hepatocyte expansion
.
Previous reports have also shown that, upon induction by small molecules, mouse and human PH can be transformed into tubular hepatic progenitor cells (LPCs) that exhibit the potential for cell proliferation and differentiation into functional hepatocytes
.
However, some disadvantages remain, including immune responses between allografts and ethical concerns
.
Schematic diagram of the article (figure from Hepatology) Therefore, gene therapy of autologous reprogrammed hepatocytes from WD animals or patients becomes a potential option
.
Previous data demonstrated the feasibility of lentivirus-mediated ATP7B overexpression in iPSC-differentiated HLC from WD patients or ATP7B gene transfer into bone marrow mesenchymal stem cells (BM-MSCs) from LEC rats
.
Both have been shown to reduce copper load in cells or in rat livers
.
To achieve this, PH from ATP7B −/− mice was reprogrammed to generate LPC by modifying an earlier established reprogramming protocol
.
This study found that hepatocyte reprogramming and expansion medium (HREM) significantly promoted LPC expansion to more than 40 passages
.
Following ATP7B overexpression, redifferentiated LPC-ATP7B-Heps were able to repopulate the livers of ATP7B −/− mice and reduce hepatic copper accumulation
.
Thus, LPC-ATP7B-Heps show therapeutic potential, and the results could pave the way for the development of new treatments for WD and similar genetic diseases of the liver
.
Reference message: https://aasldpubs.
onlinelibrary.
wiley.
com/doi/10.
1002/hep.
32484